Here we list frequently asked questions on medical device registration in China:
- Q1. Is the product a medical device?
- Q2. How to classify your medical device product?
- Q3. Can medical device trading company apply for the registration certificate from CFDA?
- Q4. How to find the appropriate legal representative in China?
- Q5. What is the registration type?
- Q6. What is the practical operation to register your product in China?
- Q7. What is the registration process and timeline for medical device registration in China?
- Q8. What is the data requirement for medical device registration?
- Q9. What are the post-registration obligations for the applicant after approval of medical device registration certificate?
-
Q10. How to skip or reduce the clinical trial mandate?
If you product meets the definition of medical device under CFDA regulations, it is managed as medical device and there are CFDA regulatory requirements that apply. The definition of medical device is as fallow:
"Medical devices" refers to: any instrument, apparatus, appliance, material, in vitro diagnostic reagents and calibration substances and other similar substances and related articles, including the needed computer software. Its main effectiveness is achieved via physics ways and so on. It does not achieve its principal action in or on the human body by means of pharmacology, immunology or metabolism, but which may be assisted in its function by such means; the use of which is to achieve the following intended objectives:
- Diagnosis, prevention, monitoring, treatment or alleviation of disease;
- Diagnosis, monitoring, treatment, alleviation of or compensation for an injury or handicap conditions;
- Investigation, replacement or modification for anatomy or a physiological process;
- Life support or maintenance;
- Control of conception;
- Offer information for medical or diagnosis purpose via inspecting the human samples.
back to top
Q2. How to classify your medical device product?
Medical Devices (including IVD) are divided into three managing categories: class I, class II and class III medical devices based on the different risk level. There are higher controlled requirements if the product classified as higher level, such as class II or class III, and there are different requirements for each level as well.
The classification assigned based on risk and should be determined by the combined judgment on three aspects: intended purpose, structural features, intended for use and use descriptions. You can classify your product according to the guideline on medical device classification.
1. Search the medical device classification catalogue or database
China Food and Drug Administration (CFDA) has identified the classifications for approximately 4,000 different generic types of devices and grouped them into 44 medical specific categories. Each of these generic types of devices is assigned to one of three regulatory classes based on the level of control necessary to assure the safety and effectiveness of the device.
You can determine the regulatory class for you product by matching your product with the device listed in the medical device classification catalogue or database.
If you hope to search the English version of medical device classification catalogue, please contact Mr. Wen (Edwin.wen@cirs-group.com).
2. Classify your product based upon the classification rules
The classification of medical device is determined by the combined judgment from four aspects: intended purpose, structural features, intended for use and use descriptions. You can determine the regulatory class for your product by screening these factors by yourself or using CIRS Medical Device Classification System.
3. Apply for classification determination from CFDA
If you product is unclassified or difficult to determine the classification based on existing knowledge, especially the medical devices contained drug, disinfectant or cosmetic, you can apply for classification determination from CFDA via CIRS.
back to top
Q3. Can medical device trading company apply for the registration certificate from CFDA?
No, medical device trading company can’t apply for the registration certificate from CFDA, there are only medical device manufacturer qualified to act as applicant under CFDA regulations. Foreign manufacturer should appoint a Chinese legal representative to handle the regulatory affairs in China. This is mandatory if you have no China office.
The regulatory obligations of your Chinese legal representative should be as follow:
- Contact with the corresponding food and drug administration department and the foreign applicant (or proposer).
- Convey the regulations and technical requirements to the foreign applicant (or proposer) accurately and precisely.
- Collect adverse events of the post-marketing medical device and feed back to the foreign applicant (or proposer), and report it to the corresponding food and drug administration department.
- Recall the post-marketing medical device which has quality matters and report it to the corresponding food and drug administration department
- Other responsibility related to product quality and after-sale service.
Q4. How to find the appropriate legal representative in China?
Your local legal representative must have a valid business registration, full range service and local residence in China. You can appoint your China subsidiary office or a qualified company as your legal representative. The authorized company should have a sufficient background in the practical handling of regulatory affairs of medical device in China.
back to top
Q5. What is the registration type?
| Medical Devices | Approval | Testing | Clinical Trial | Authorities | Deadline |
| Class I | Record | Self-testing | N/A | CFDA | Pre-market |
| Class II | Initial Registration | Required | Required | CFDA | Pre-market |
| Updates | TBA | TBA | CFDA | Within 1months after being updated | |
| Renewal | TBA | N/A | CFDA | 6 months before the date of certificate expired | |
| Class III | Initial Registration | Required | Required | CFDA | Pre-market |
| Updates | TBA | TBA | CFDA | Within 1months after being updated | |
| Renewal | TBA | N/A | CFDA | 6 months before the date of certificate expired |
Q6. What is the practical operation to register your product in China?
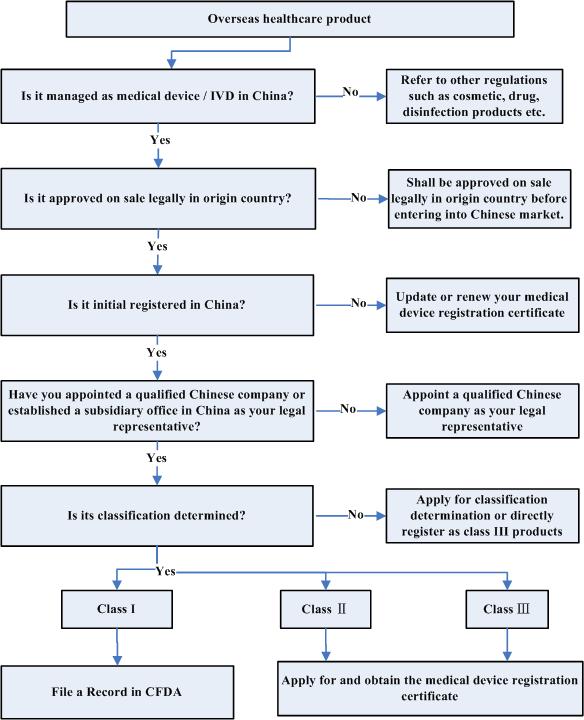
back to top
Q7. What is the registration process and timeline for medical device registration in China?
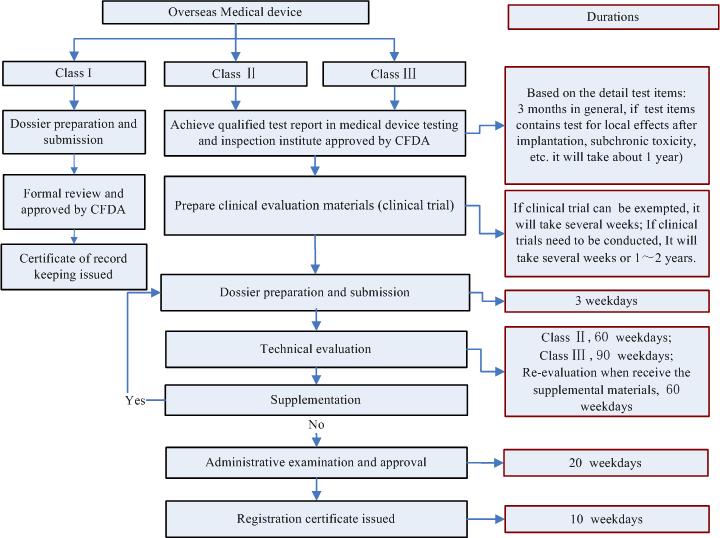
back to top
Q8. What is the data requirement for medical device registration?
| level I headlines of application dossier | level II headlines of application dossier |
| 1. Application form | |
| 2. Supporting documents | |
| 3. List of basic requirements for safety and effectiveness of medical device | |
| 4. Summary |
4.1 Overview 4.2 Product description 4.3 Model and specification 4.4 Packing instructions 4.5 Applicable scope and contraindications 4.6 Referenced clinical data of products of same kind or its predecessors (if any) 4.7 Other contents to be specified |
| 5. Research Information |
5.1 Product performance study 5.2 Evaluation study on biocompatibility 5.3 Biological safety study 5.4 Sterilization / disinfection process study 5.5 Study on shelf life and packaging 5.6 Animal study 5.7 Software study 5.8 Others |
|
6. Manufacturing information |
6.1 Information description of the manufacturing process of non-active/active products 6.2 Manufacturing site |
| 7. Clinical evaluation material | |
| 8. Risk analysis material of products | |
| 9. Product technical requirement | |
| 10. Product Type-testing report |
10.1 Type-testing report 10.2 pre-assessment advice on Product technical requirement |
| 11. Samples of instruction for use and label |
11.1 Instruction for use 11.2 Label samples of minimum sales unit |
| 12. Conformity statement |
Q9. What are the post-registration obligations for the applicant after approval of medical device registration certificate?
Medical devices manufacturer as well as other firms involved in the distribution of medical devices must follow the appropriate requirements and regulations once their product approved by CFDA and placed on Chinese market. The post-market obligations in China shall include inspection and supervision, advertisement approval, adverse events monitoring, clinical data gathering and reporting, quality management system stewards, product recall and annual reporting.
back to top
Q10. How to skip or reduce the clinical trial mandate?
Being in accordance with clinical trial requirements and associated submission processes under the new CFDA regulations, foreign medical device manufacturer are required to conduct the clinical trial in a registration process for the higher risk products, those designated as class II or III under the CFDA medical device classification system. You can eliminate or reduce the clinical trial when meet the following situations:
1. Your product has been listed on “the catalogue of medical device exempted from clinical trial” exactly, it will be exempted from clinical trial requirements and only required to submit the clinical evaluation report to identify the same technical features in comparing with the medical device listed on the catalogue.
2. Your product may be exempted from clinical trial requirements when meet any one of the following conditions:
a). With explicit working mechanism, complete design, skilled manufacturing technique, or similar products that have been working on the market for several years and with no
serious adverse events and the general-purpose doesnt change.
b). The safety and effectiveness of medical device can be ensured via other data rather than clinical trials.
c). The safety and effectiveness of medical device can be ensured by analyzing and evaluating the data gained from clinical trials or the data of same type medical devices.3. You
can use the clinical evidence reports to reduce the size of or eliminate the clinical trials, the clinical study report with clinical process and data which approved in origin
authorities would be the best material for clinical evaluation report preparation, in additionally, please gather and prepare the analysis documents for race difference, epidemic
disease background, regional disparity, etc.
back to top






